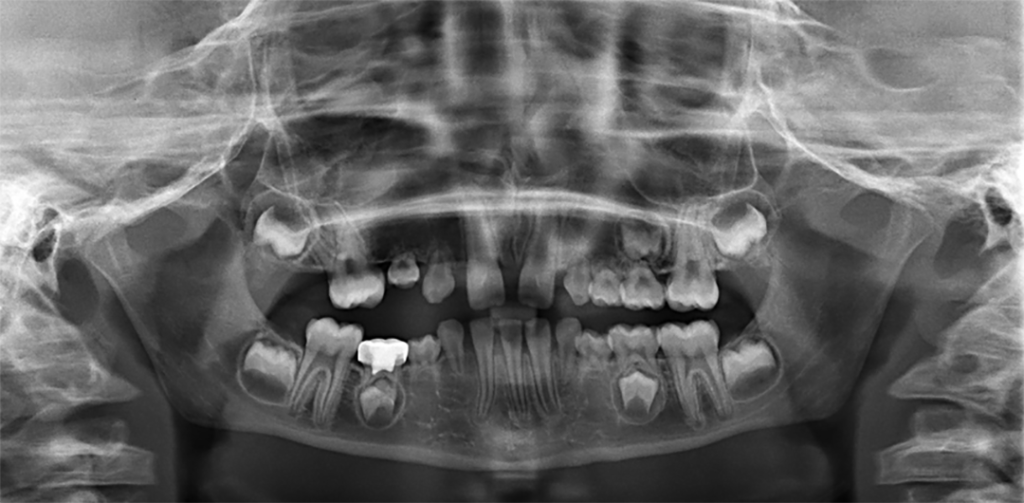
Osteogenes imperfecta (OI) é uma doença hereditária do tecido conjuntivo com muitas apresentações associadas a fatores adquiridos pelo ambiente. Frequentemente, é chamada de “doença dos ossos quebradiços” e os pacientes gravemente afetados sofrem fraturas múltiplas, com mínimo ou nenhum trauma, e bebês com a pior forma de OI morrem no período perinatal. As formas leves dessa condição podem se manifestar apenas com osteoporose prematura ou perda mineral óssea grave na pós-menopausa.
A causa da OI é estabelecida na maioria dos casos. Em pacientes com defeitos moleculares identificados, a osteogênese é mais comumente causada por mutações em genes que codificam as cadeias alfa 1 e alfa 2 do colágeno tipo I ou proteínas envolvidas na modificação pós-tradução do colágeno tipo I. A qualidade óssea defeituosa explica muitos aspectos clínicos dessa doença.
OI é classificada em uma série de subtipos principais com base em características genéticas, radiográficas e clínicas. Vamos conhecer a divisão clínica mais útil, baseada nos problemas típicos que se manifestam em bebês, crianças e adultos com doença leve, moderada a grave e letal?
As manifestações clínicas variam substancialmente dentro das famílias:
Para os tipos mais leves de OI, um membro pode ser significativamente afetado clinicamente, enquanto outro membro com a mesma mutação pode ter função normal. Isso enfatiza que a identificação de uma mutação em um determinado gene não resulta necessariamente em um diagnóstico clínico claro, e sugere que pode ser necessário defeitos em outros componentes do tecido conjuntivo para expressar totalmente essa síndrome genética.
As manifestações clínicas de OI incluem:
●Excesso ou fraturas atípicas (ossos quebradiços): as fraturas mais comumente associadas com OI foram as fraturas transversas do úmero, olécrano e diafisária do úmero;
●Baixa estatura;
●Escoliose;
●Deformidades do crânio basilar, que podem causar compressão do nervo ou outros sintomas neurológicos;
●Esclera azul;
●Perda auditiva (geralmente detectada no final da infância até o início da idade adulta);
●Dentes opalescentes que se desgastam rapidamente (dentinogênese imperfeita);
●Aumento da flacidez dos ligamentos e da pele;
●Ossos wormianos (ossos pequenos e irregulares ao longo das suturas cranianas);
●Possibilidade de formação de hematomas com facilidade.
Ela ainda pode ser dividida em tipos de acordo com os seus sintomas:
Leve (tipo I) – A fragilidade óssea é a menos severa na OI tipo I. A taxa de fratura é variável. Esses indivíduos podem ter poucas ou nenhuma fratura antes da puberdade, ou numerosas fraturas ao longo de suas vidas. A deformidade é mínima e a estatura geralmente é normal;
Forma perinatal letal (tipo II) – Pacientes com OI perinatal letal (tipo II) geralmente morrem no útero ou na primeira infância. Fraturas graves e insuficiência pulmonar são problemas típicos que aceleram a morte nesse grupo. O aconselhamento genético é indicado para famílias afetadas. O tratamento da OI tipo II é de suporte;
Moderada a grave (tipos III a IX, XV e XVI) – A fragilidade óssea é moderada a grave em pacientes com OI tipos III a IX, XV e XVI. Aqueles com OI tipo III são geralmente os mais gravemente afetados. No entanto, crianças com OI tipo VII e VIII, ocasionalmente desenvolvem um tipo grave e letal de OI semelhante à OI tipo II.
O diagnóstico clínico da osteogênese imperfeita é baseado nos sinais e sintomas descritos acima. A definição da doença, geralmente, é direta em indivíduos com fragilidade óssea e uma história familiar positiva ou várias manifestações extraesqueléticas. No entanto, na ausência dessas características, o diagnóstico pode ser difícil. Não há teste de laboratório definitivo e prontamente disponível para OI.
O tratamento consiste em reduzir as taxas de fratura, prevenir deformidades de ossos longos e escoliose, minimizar a dor crônica e maximizar a mobilidade e outras capacidades funcionais. Pacientes com OI devem ser encaminhados para avaliação por especialistas em genética e, se clinicamente indicado, ao ortopedista com experiência no tratamento dessa condição. O tratamento requer uma abordagem de uma equipe multidisciplinar coordenada e consiste em fisioterapia, intervenções cirúrgicas e medicamentos. Indivíduos com essa síndrome genética precisam de supervisão de saúde adicional de seus prestadores de cuidados primários e monitoramento de complicações potenciais.